▶第36号(2008年 3月1日発行)
多発性嚢胞腎
標本館運営委員 舘 延忠
札幌医科大学保健医療学部 作業療法学科准教授
これは(図1)標本館に展示されている腎不全で死亡し病理解剖をうけた多発性嚢胞腎(polycystic kidney disease : PKD)のマクロの病理標本である。1976年に標本館に展示された。病理解剖の記載によると生前の診断はPKDで、両側のPKDと多発性肝嚢胞も認められた。この機会にPKDに関して概説してみたい。
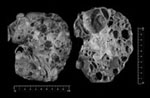
図1. 多発性嚢胞腎の剖検腎. 腎は著しく腫大し多発性の嚢胞を認める. ホルマリン固定.
病理学的にPKDは、両側腎のネフロンと集合管に嚢胞が生じ次第に数と大きさが増大し、腎機能が低下する疾患と定義される。先天性と後天性に大きく分類され、多くは先天性である。最近の分子遺伝学の進歩により原因遺伝子が同定されてきた。先天性の中でも常染色体優性多発性嚢胞腎(autosomal dominant polycystic kidney disease : ADPKD)は1000人に1人と、遺伝子腎疾患者の中で最も頻度が高く、従来、成人型多嚢胞腎と呼ばれてきた。しかし、近年、学校検尿にて蛋白尿、血尿を契機に小児期にCTなどにより診断されることもある。ADPKDは、両側性の腎嚢胞、肝臓、膵臓、くも膜の嚢胞、脳動脈瘤や大動脈瘤などの血管異常を伴う。多発性肝嚢胞はADPKDに最も高頻度にみられる腎外病変で50歳以上のADPKDの患者の75%に認められる。原因遺伝子として16p13.3に存在するPKD1遺伝子(遺伝子産物はpolycystin-1)の異常によるものが85%と頻度が高く、4q13-q23に存在するPKD2遺伝子(遺伝子産物はpolycystin-2)の異常によるものが15%である。
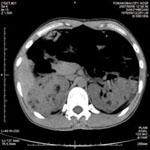
図2. 腎臓のCT. 両側の腎臓に多発性の嚢胞を認める. 結節性硬化症の症例.
polycystin-1は尿細管の成熟過程で発現し尿細管細胞の接着と分化に関与している。polycystin-2は細胞内小器官に存在しpolycystin-1と相互作用し細胞内Caを調節しているので、このいずれかの異常が相互作用を破綻することでADPKDが発症すると考えられている。遺伝子型と臨床所見との相関をみると、腎不全になる平均年齢はPKD1異常では53歳、PKD2異常では69歳とPKD2が軽度である。平均すると60歳までに約50%前後が腎不全になる。ここに提示された症例に関しては、この当時はCTはなく、生前のPKDの診断は詳細な臨床検査によってなされた。多発性肝嚢胞を有してよりADPKDの可能性が高い。近年、エコー、CT(図2)、MRIなどの画像解析の導入により、多発性嚢胞腎の診断は容易になった。ADPKDの確定診断は腎の画像診断でなされ、50%のリスクがある人に対する診断基準は1)30歳未満で、少なくとも片側性もしくは両側性の嚢胞が2個以上ある。2)30-59歳で、両側の腎にそれぞれ2個の嚢胞がある。3)60歳以上で、両側の腎にそれぞれ4個の嚢胞がある。この診断基準の感度は30歳以上のADPKD患者や30歳未満でPKD1遺伝子変異をもつ患者に対しては100%である。症例によって画像診断を含めた臨床診断が不確実な場合や家族歴がない時には、確定診断のためにPKD1およびPKD2遺伝子の解析が必要である。また遺伝子解析により出生前診断も可能になったが、成人で発症、ある程度の治療法もあるので、現実的にはやられていない。ホルマリン長期保存臓器からの遺伝子抽出が可能になり、この症例の剖検ホルマリン臓器からPKD1およびPKD2遺伝子を解析することでADPKDの確定診断は可能である。頻度は少ないが、その他の先天性PKDとしては、常染色体劣性多発性嚢胞腎(autosomal recessive polycystic kidney disease : ARPKD)がある。常染色体劣性遺伝形式であるより頻度は少ないが、ADPKDに比べ重症である。新生児から乳児期に腎不全になり死亡する症例が多い。6p12-21.1に存在するpolyduction/fibrocystin蛋白をコードするPKHD1遺伝子のホモ接合変異で発病する。両側腎の集合管の拡張により、腎臓は腫大し多くは、出生時から腎機能障害を認め、腎不全になる。新生時から腎不全を示す例はPotter症候群と呼ばれ、高度の腎形成障害による妊娠中の羊水減少、出生後の肺低形成による呼吸不全を伴い予後不良である。その他小児科領域では、結節性硬化症(tuberous sclerosis)に伴うPKDがみられる。ここに提示した多発性嚢胞腎のCT(図2)は結節性硬化症の患者さんである。本人の同意を得て提示させていただいた。脳、心臓、腎臓などの臓器に過誤腫性病変を伴う神経皮膚症候群の1つで、精神遅滞、難治性てんかん、皮膚症状を主症状とし常染色体優性遺伝形式をとるが、大部分は突然変異(de novo)である。皮膚症状としては顔面の血管線維腫、躯幹および四肢の白斑が特徴である。中枢神経では、大脳皮質および皮質下の多発性の結節を伴い、それによるてんかんと知能障害を認める。皮質下結節は石灰化を伴うより脳CTで脳室周囲に石灰化を認めれば、皮膚の白斑と併せて乳幼児期での、結節性硬化症の早期診断ができる。腎臓に関しては、血管筋脂肪腫やPKDが高頻度で認められ、PKDは幼少時ではみられないが、学童期以降に20%の症例に見られる。結節性硬化症の原因としては癌抑制遺伝子である9q34に存在するhamartin(TSC1)または、16p13に存在するtuberin(TSC2)遺伝子異常で結節性硬化症の患者の80%に認められる。また、ADPKDの原因遺伝子PKD1とTSC2は16p13にあって近接しているより、この部分の欠失は早い時期よりPKDを認める結節性硬化症になる(TSC2/PKD contiguous gene syndrome)。またTSC2変異を持つ患者さんは、TSC1変異を持つ患者さんより重症であり腎病変の頻度が高い。臨床所見、CTおよびMRIで結節性硬化症の診断はされるが、非定型例などでは遺伝子診断が有用である。
以上、多発性嚢胞腎のなかで成人と小児に頻度の高い病型を挙げて臨床および分子遺伝学的な観点より概説してみた。
文 献
1) Guanqing W. Current advances in molecular genetics of autosomal-dominant polycystic kidney disease. Curr Opin Nephrol Hypertensions 2001; 10: 23-31.
2) Hanaoka K, Qian F, Boletta A, etal. Co-assembly of polycystin-1 and -2 products unique cation-permeable currents. Nature 2000; 408;990-994.
3) Bear JC, Parfrey PS, Morgan JM, et al. Autosomal dominant polycystic kidney disease: new information for genetic counseling. Am J Med Genet 1992;43:548-553.
4) Bear JC, McManamon P, Morgan J, et al. Age at clinical onset and at ultra-sonographic detection of adult polycystic kidney disease: data for genetic counseling. Am J Med Genet 1984; 18: 45-53.
5) Lamb RF, Roy C, Diefenbach TJ, et al. The TSC1 tumor suppressor hamartin regulates cell adhesion through ERM proteins and the GTPase Rho. Nat Cell Biol 2000; 2: 281-287.
6) 菊池 真、舘 延忠ら. ホルマリン固定組織からのDNA抽出法とPCR法による遺伝子解析. 札幌医学雑誌 2005; 74; 39-44.